
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
索托拉西布通过FABP4-PPARγ-CPT1B轴诱导线粒体氧化应激导致间质性肺病的新型脱靶机制研究
【字体: 大 中 小 】 时间:2025年10月04日 来源:Cell Communication and Signaling 8.9
编辑推荐:
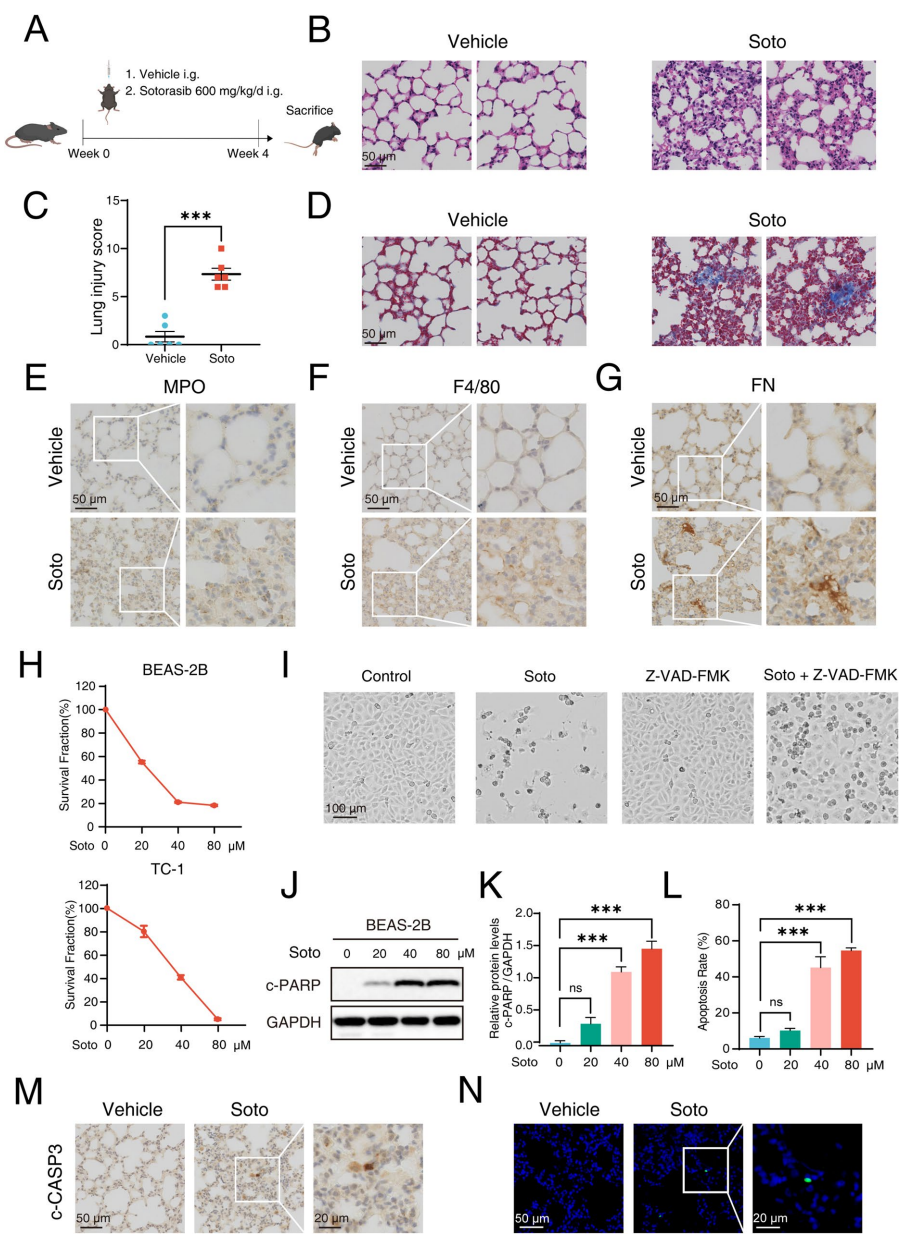
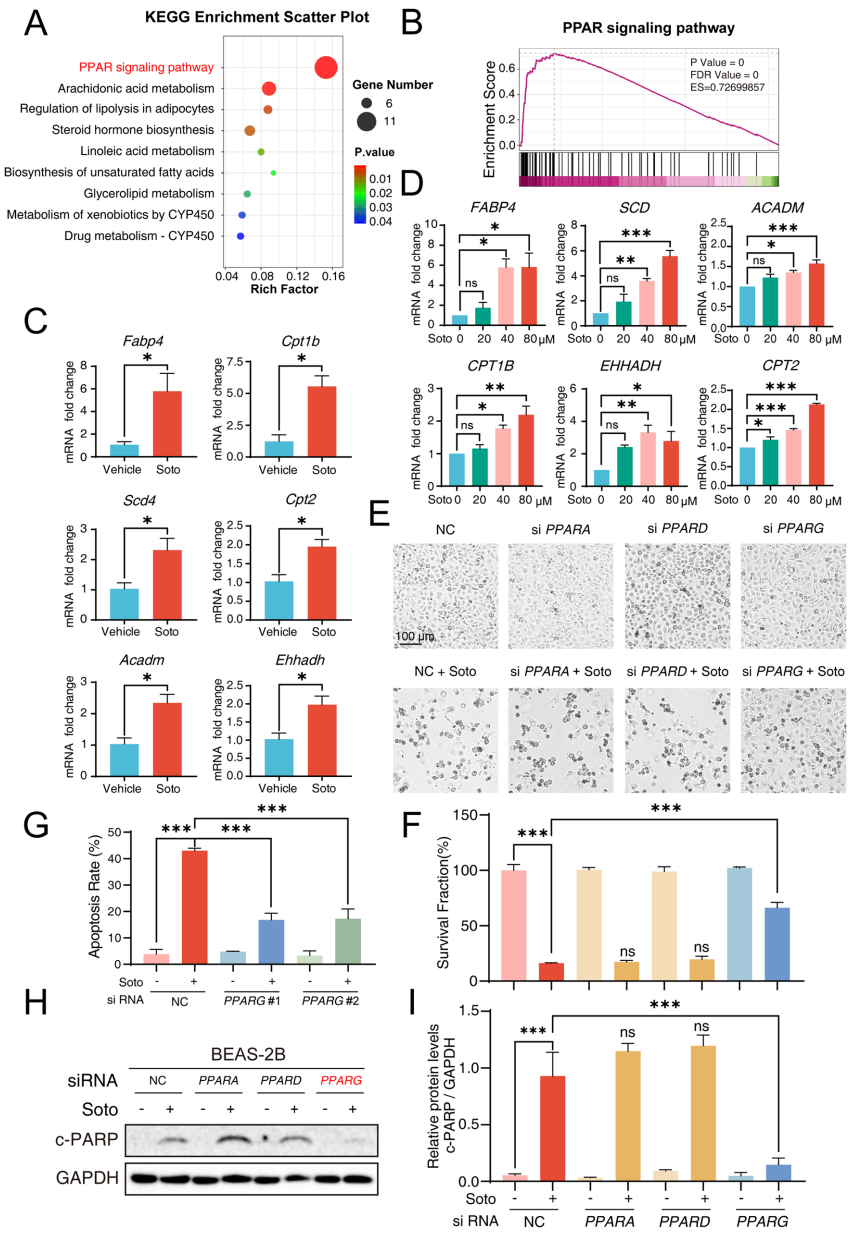
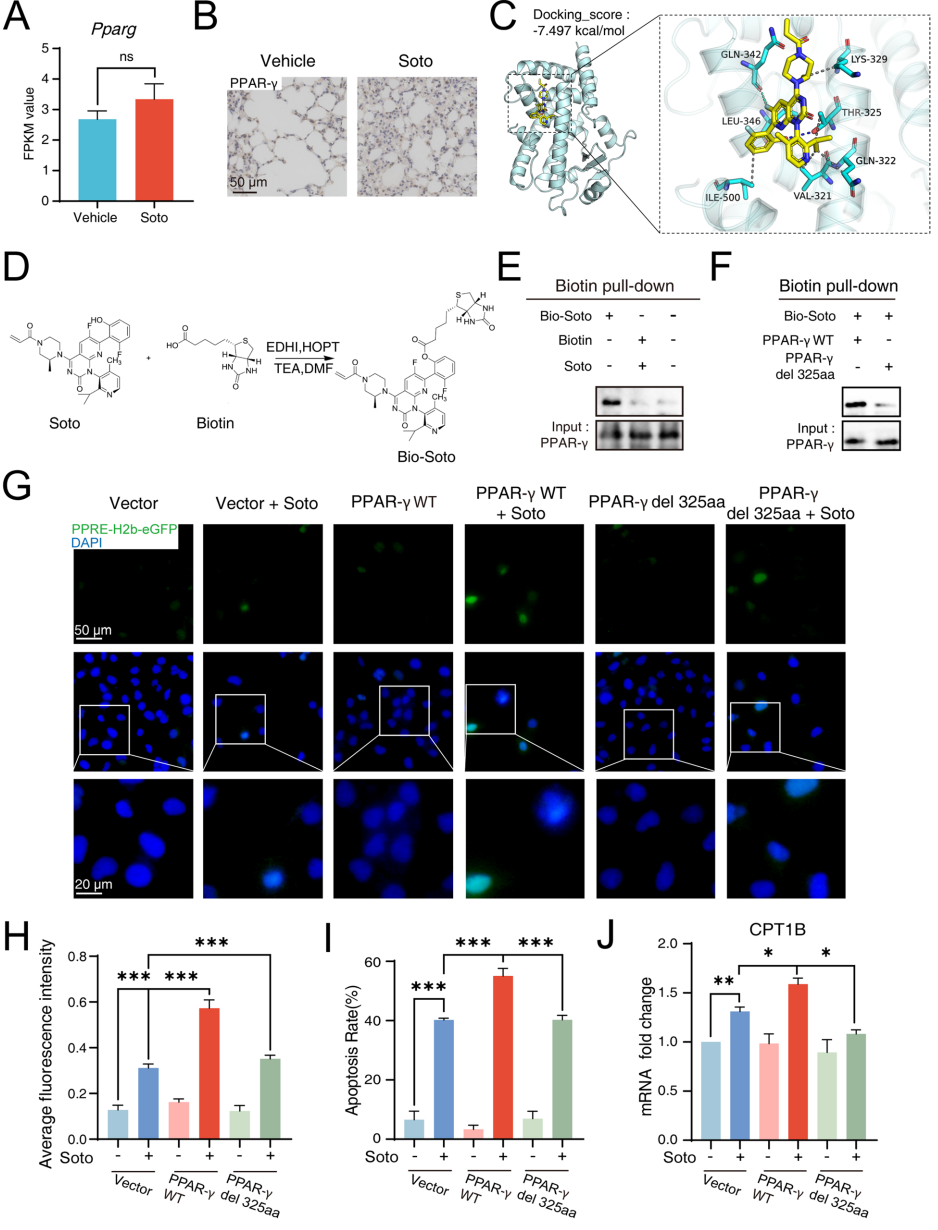
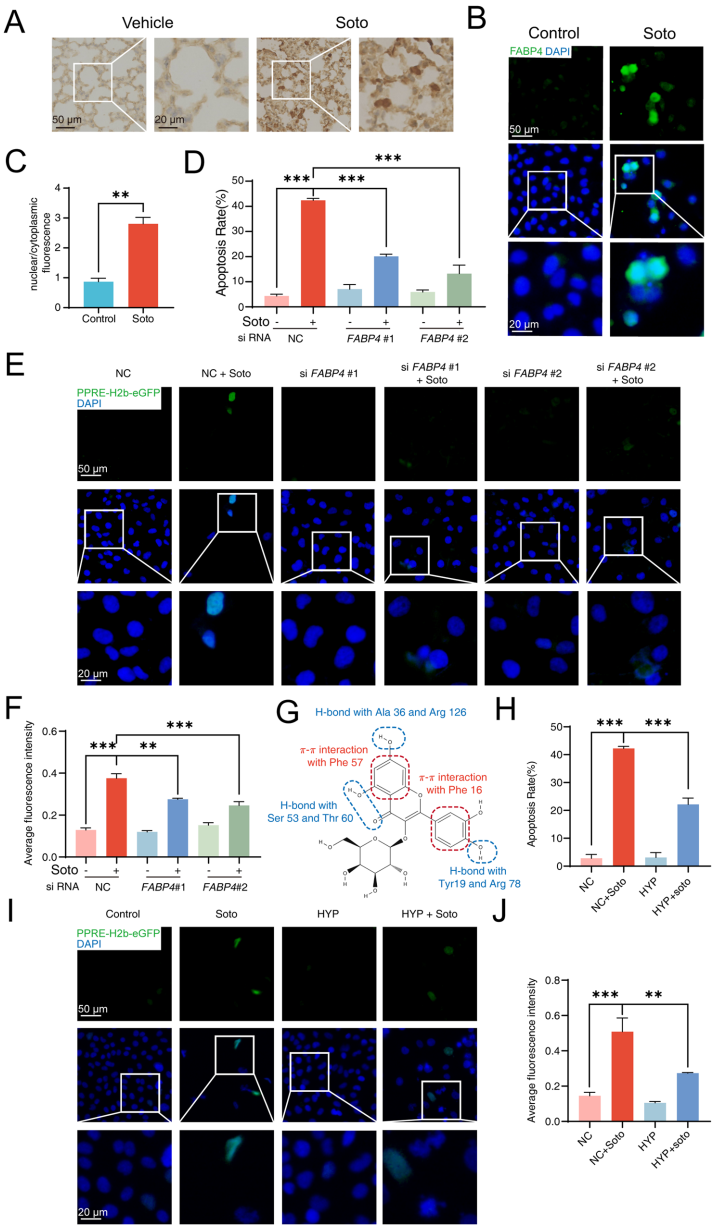
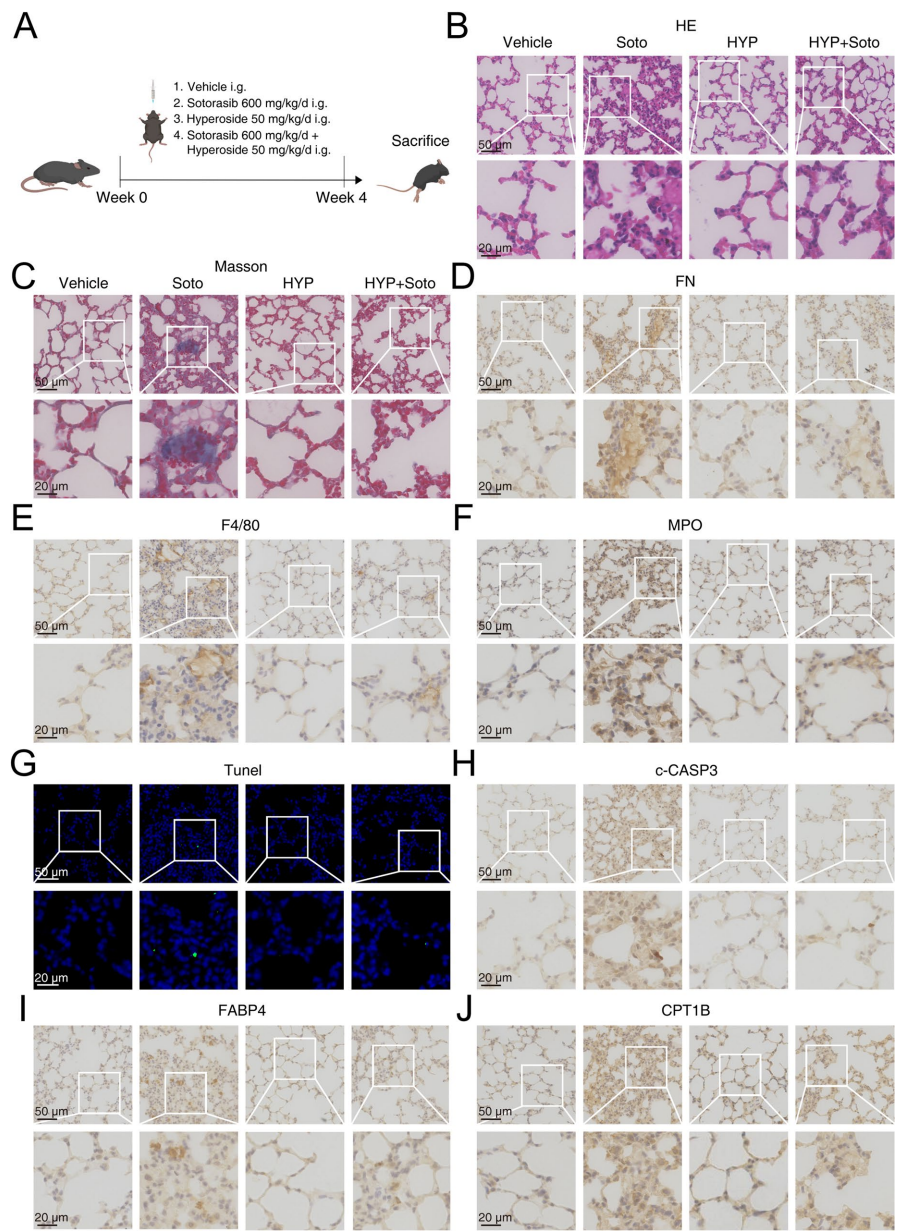
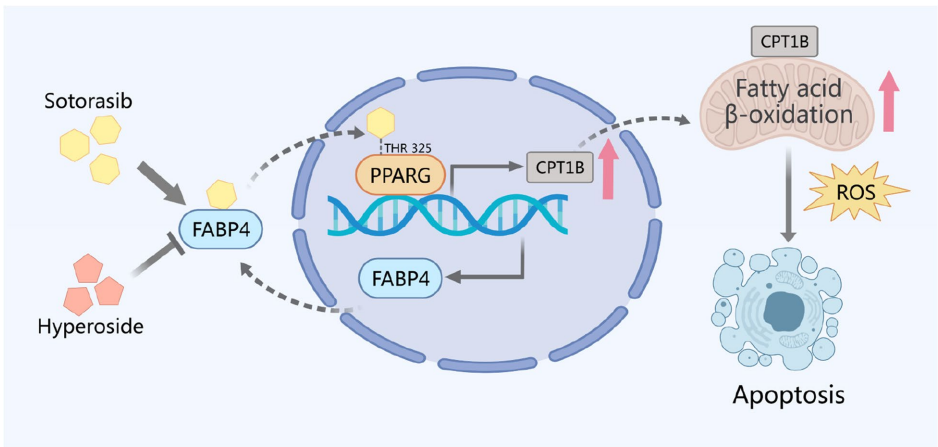
本刊推荐:为解决KRAS G12C抑制剂索托拉西布(sotorasib)临床应用中引发的致死性间质性肺病(ILD)机制不明问题,研究人员开展药物脱靶效应研究,发现sotorasib通过结合PPARγ Thr325位点,经FABP4介导核转位激活CPT1B依赖性脂肪酸氧化(FAO),导致线粒体ROS过量产生并诱发肺泡上皮细胞凋亡及纤维化。该研究揭示FABP4抑制可成为临床干预新策略,为增强靶向药物安全性提供重要依据。
 生物通微信公众号
生物通微信公众号
知名企业招聘

