
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
综述:细胞焦亡在卵巢癌中的作用
【字体: 大 中 小 】 时间:2025年10月05日 来源:Hormones & Cancer
编辑推荐:
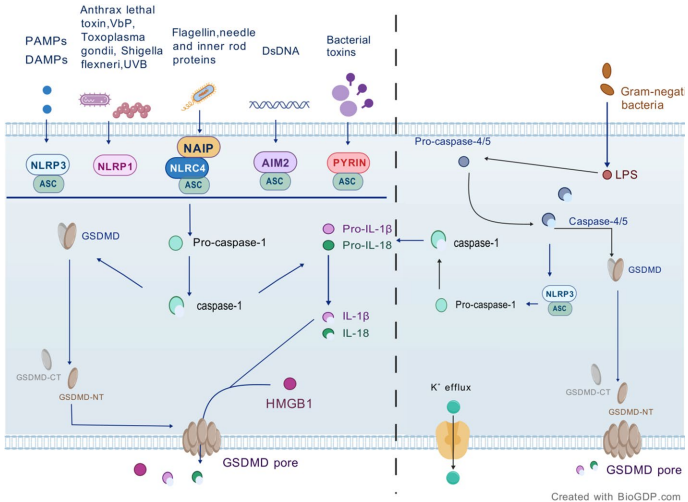
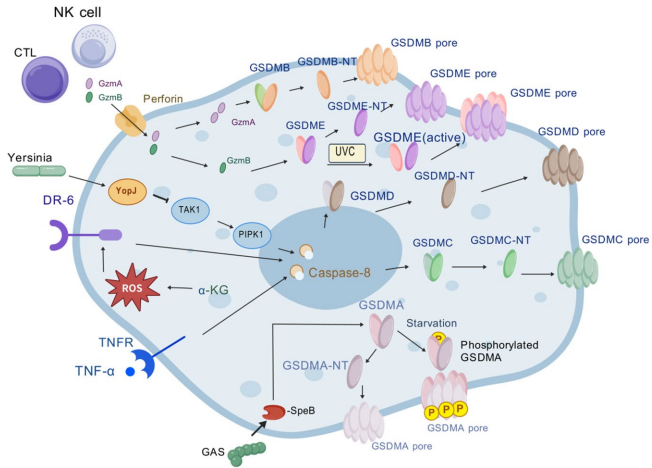
本综述系统探讨了细胞焦亡(pyroptosis)的分子机制及其在卵巢癌(OC)发生、发展、预后和治疗中的作用。文章深入解析了炎症小体(inflammasome)、半胱天冬酶(caspase)和Gasdermin(GSDM)家族蛋白介导的经典与非经典通路,强调了其在肿瘤免疫微环境(TME)调控和化疗耐药中的双重角色,为卵巢癌的监测与治疗提供了新视角。
 生物通微信公众号
生物通微信公众号
知名企业招聘

