�ӻ������źŵ�Ԥ��ߣ��ɾ���-����Ԥ��Ĺؼ������������ϸ������
��Briefings in Bioinformatics����From genomic signals to prediction tools: a critical feature analysis and rigorous benchmark for phage�Chost prediction
�����壺
��
��
С
��
ʱ�䣺2025��11��25��
��Դ��Briefings in Bioinformatics 7.7
�༭�Ƽ���
�������о���Բ���-���������Ԥ���������������һ�������Բ�������״��ϵͳ�ع˲��ϸ�������27��Ԥ��ߡ��о���Ա������RefSeq-VHDB��MetaHiC-VHDB���������ݼ�������CHERRY��iPHoP�ȹ��߾��й㷺�����ԣ���RaFAH��PHIST���ض������������죬��ʾ��Ԥ��ȷ�ԡ�Ԥ���������ɱ�֮��Ĺؼ�ƽ�⣬Ϊ�о���ѡ�ߺ��ƶ����������ṩ��ʵ��ָ�ϡ�
����
���������������ս���У��ɾ�����Ϊ��������ḻ������ʵ�壬��ϸ����������������ͣϢ�Ľ���������������Щ����������������ò�������������Ⱥ�����̬�ṹ�����ǿ���Ⱦ���Ƶ���ϣ�������ɾ����Ʒ�����Ϊ�Կ���������ҩ�Ե�����������Ȼ����ʵ�������Щ����üȺ�ʱ����������������Ҫ�����Ÿ�ͨ���������ı���ʽ��չ������Ԥ�ⷽ��Ӧ�˶�����
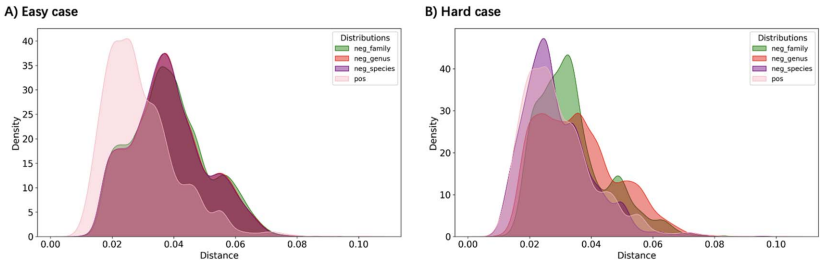
��������֮������Խ��Խ���Ԥ��߹�����һ�����ӵļ�����̬��������������һ�º��߿����Բ��죬ֱ�ӱȽ����ǵ����ܱ���쳣���ѡ������ֵ��ǣ��������ݿ�������ص�ע��ȱ�ں��о�ƫ����������������ȱ��������Ϣ������֪��������ָ߶ȼ���������ģʽ�����ϡ����⣬����������Χ��ʵ�ʸ�����Զ�����ݿ�ġ�һ��һ����ϵ�������ɾ����ܹ���Ⱦ������������������������Ԥ��ģ�͵Ĺ��������˾���ս��
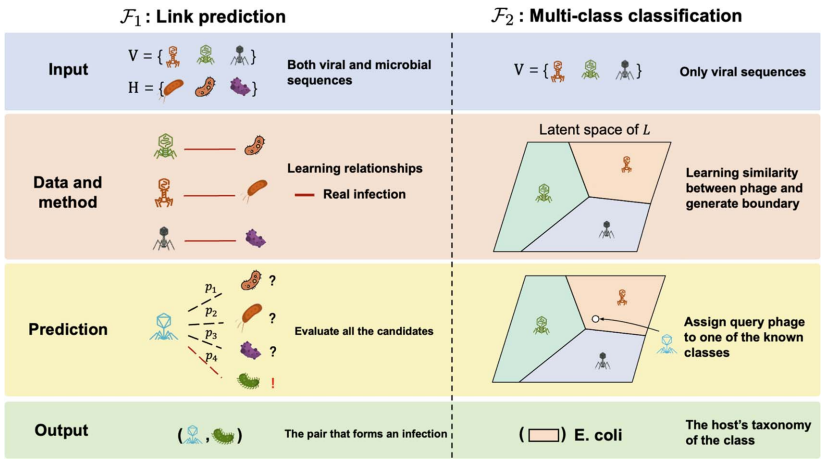
Ϊ��������һ���Ҿ��棬��۳��д�ѧ��������Ĵ�ѧ���о��Ŷ��ڡ�Briefings in Bioinformatics���Ϸ������ذ��о�����27�ֲ���-����Ԥ��߽�����ϵͳ�������ϸ��������������Ƚ�����Ԥ��������ȷ����Ϊ�����ܣ�����Ԥ�⣨link prediction���Ͷ�����ࣨmulti-class classification����������������ר����ƵĻ����ݼ�����RefSeq-VHDB�����ݿ������ͣ���MetaHiC-VHDB��������鷢���ͣ�����ģ�ⲻͬ���о�������
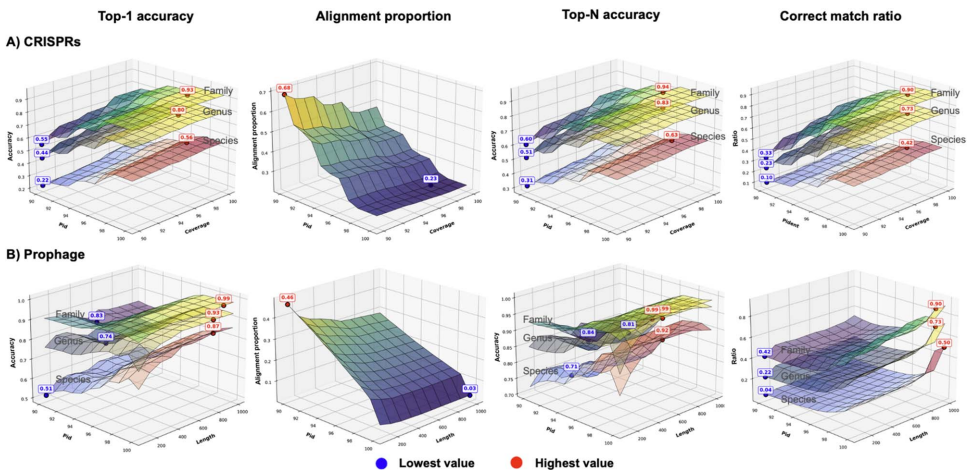
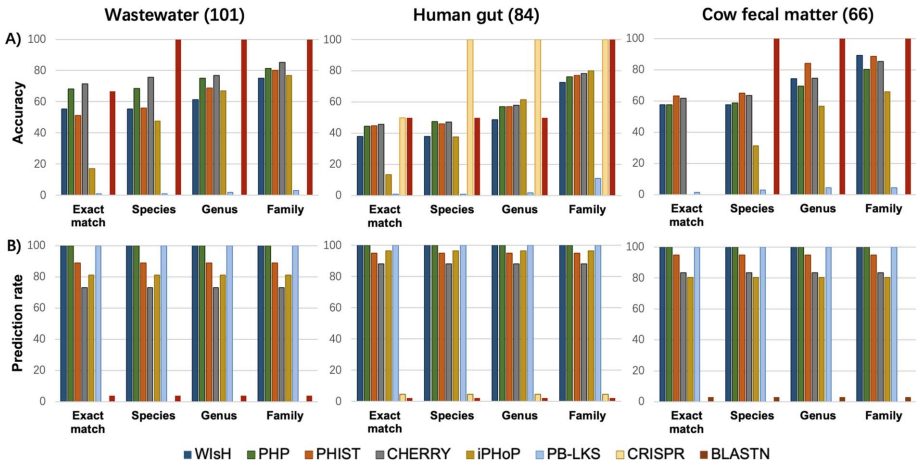
�о��ŶӲ����˶�ά���������ԣ�����ϵͳ������CRISPR�������ƥ�䡢ǰ�ɾ����⡢k-merƵ�������Եȹؼ�������������Ԥ��Ч�ã�ͨ����������ȷ���˸���������Ѳ�����ֵ�����ǹ��������������ݼ���RefSeq-VHDB����4,698���������ּ�����ע�͵IJ������У�MetaHiC-VHDB���������������೦����ţ��ͷ�ˮ������251��ͨ��Hi-C������֤�IJ���-���������ԡ�����ָ�����ȷ�ʡ�Ԥ�����Լ�����Ч�ʣ����Կ��ù��߽�����ʵ�ʰ�װ���ԡ�
�о���Ա���������˸�������ѧ����������Ԥ���еı��֡�CRISPR�������ƥ����Ϊֱ�ӻ���֤�ݣ����ϸ���ֵ�£�����һ���ԡ�98%���ܴﵽ82%������ȷ�ʣ�����Ԥ�⸲�������ޡ�ǰ�ɾ�������95%����һ���Ժ�500bp���볤�������±�����ѣ�����CRISPR����ʶ����Ļ������ص��Ⱥܵͣ����������ַ������л����ԡ�
k-merƵ�ʷ�����ʾ��4-mer��������Ч����ԶԵ����������������ͬһ���ڲ�ͬ����ʱ�������ޡ����������������Է������ڡ����������Ⱦ����������ļ��裬�����������ݿ�ע�������ԡ�����46%�IJ���������ȷ����ע�ͣ�����73%���ֳ�����������һ���ԡ�
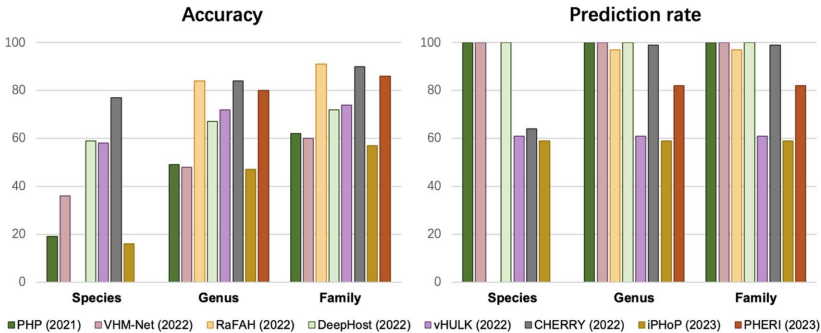
�ڽ�ʹ�ò������е�RefSeq-VHDB�����У�CHERRY������ˮƽ�������ţ�77%ȷ�ʣ���RaFAH�������ͿƼ�Ԥ�������ȣ�84%��92%����������Ԥ�����ϳ��������ֻ���PHP��VHM-Net��DeepHost�ﵽ100%Ԥ���ʣ���vHULK��CHERRY�ȹ�����ͨ���ڲ����Ŷ���ֵ���������ʱ�֤ȷ�ԡ�
��ģ����ʵ������������MetaHiC-VHDB�����У�PHP��PHIST��CHERRY������Ϊ�Ƚ���ֱ��֤�ݷ�����CRISPR��ǰ�ɾ����⣩��������Ԥ��������ȷ�ʽӽ�100%����Ԥ���ʲ���5%���������ڸ������ϵľ����ԡ�
�о������֣����á�����+��ʶ����joint+ consensus�����ɲ����ܽ�RefSeq-VHDB�ϵ�ȷ��������99%����������Ԥ�����������͡�����Ч��������ʾ��������PHIST��PHP���費��1���Ӵ���1000���������У���iPHoP��PB-LKS����Ҫ����ʱ�䣬���ֲ�����ҪԴ��������ȡ���ԵIJ�ͬ��
�����о��״�Ϊ����-����Ԥ����������ϵͳ����������ܺͱ���������Ҫ�ķ����ǣ�û�е�һ���������г����¶������ŵģ����ܸ߶������ھ���Ӧ�ó�����CHERRY���ֳ���㷺�������ԣ���RaFAH��PHIST�ȹ������ض������б���Խ���о���ʾ��ȷ�ԡ�Ԥ���ʺͼ���ɱ�֮�䲻�ɵ��͵�����ƽ���ϵ��Ϊ��ͬ������о����ṩ����ȷ��ѡ��ָ�ϡ�
�ù�������Ҫ�������ڽ���Ƭ���Ĺ��������������ƶ�������Ľ�����չ�������Ļ����ݼ������������ڷ����ȽϺʹ��£���ʵ�õĹ���ѡ��ָ�Ͻ����ٲ���-����������о�����̬ѧ��ҽѧ�����\���е�Ӧ�á��ر��Ƕ����ɾ����Ʒ�������ȷʶ��������Χ�dzɹ�Ӧ�õĹؼ�ǰ�ᡣ
δ����ս�����������ɾ����������ΧԤ�⡢���ϵ�ϸ��������¼�������̬�������̣��Լ���������չ��RNA�������������Ϊ�����������ḻ������ʵ�塪���ɾ�����������֮�临�ӵ����������춨�˼�ʵ��������־�ż��㲡��ѧ�������ʵ�û���������Ҫһ����
 ����ͨ�Ź��ں�
����ͨ�Ź��ں�
 ����ͨ������
����ͨ������
- ����
- ����
- ����
- ����
- ��ҵ
- �ȵ�
- ����
���ն�̬ |
�˲��г� |
�¼���ר�� |
�й���ѧ�� |
��չ̨ |
BioHot |
�ƽ���ֱ�� |
��չ���� |
�ؼ�ר�� |
������Ѷ |
�������
��Ȩ���� ����ͨ
Copyright© eBiotrade.com, All Rights Reserved
��ϵ���䣺
��ICP��09063491��