
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
利用宏基因组共组装与分箱策略挖掘低丰度未培养基因组实现结直肠癌高精度预测
【字体: 大 中 小 】 时间:2025年09月21日 来源:BMC Cancer 3.4
编辑推荐:
本研究针对传统物种注释方法无法检测未培养物种的局限,通过宏基因组共组装和分箱技术从亚洲和高加索人群结直肠癌队列中回收高质量基因组,发现低丰度物种在疾病分类中具有关键作用。研究人员通过随机森林特征选择构建预测模型,在两组队列中分别达到0.90和0.98的AUROC值,证实未培养物种对结直肠癌预测具有重要价值,为疾病机制研究和临床诊断提供新方向。
在人体这个复杂的生态系统里,数以万亿计的微生物与我们共生,尤其在肠道中形成的菌群网络更是影响着人类健康。近年研究发现,肠道微生物与肥胖、糖尿病、结直肠癌(CRC)、甚至神经系统疾病都存在密切关联。然而,当前主流的微生物组研究技术――例如16S rRNA测序和全宏基因组测序(WMS)结合物种注释分析――存在明显局限:它们高度依赖现有数据库,无法检测环境中大量存在的“未培养物种”(uncultivated species),即所谓“微生物暗物质”。这些未被认识的微生物很可能在疾病发生中扮演关键角色,尤其在结直肠癌这类与菌群紊乱密切相关的疾病中。
为此,Po-Ting Lin与Yu-Wei Wu团队在《BMC Cancer》发表了一项创新研究,他们采用宏基因组共组装(co-assembly)与分箱(binning)策略,从亚洲与高加索两个人群的CRC队列中成功回收了大量未培养物种和低丰度物种的基因组,并证明这些物种在区分癌与非癌样本中起到关键作用。通过机器学习建模,仅使用数十个筛选出的基因组特征即可实现结直肠癌的高精度预测(AUROC最高达0.98)。该研究不仅拓展了肠道菌群与疾病关联研究的深度,也为未来基于宏基因组的无创诊断工具开发奠定了方法学基础。
研究人员主要采用了以下几种关键技术方法:
首先,从欧洲核苷酸档案(ENA)获取亚洲(Yu等,2017)和高加索(Feng等,2015)人群的CRC宏基因组数据,排除腺瘤样本后分别进行质控与共组装(使用MEGAHIT)。随后通过MaxBin2进行基因组分箱,获取宏基因组组装基因组(MAGs),并利用CheckM筛选中等质量以上(完整度>50%,污染率<10%)的基因组。使用GTDB-tk进行物种注释与平均核苷酸一致性(ANI)分析。最后,基于基因组覆盖度进行差异分析(edgeR),并采用随机森林(Random Forest)分类与10折交叉验证评估特征重要性及预测性能。
通过共组装和分箱,研究从亚洲和高加索队列中分别获得351和458个中等质量以上的MAGs。物种分布分析显示两队列共享167个物种,其中亚洲与高加索队列分别有13.76%和11.11%的基因组因ANI值低于95%而被归类为未培养物种,包括15个共有的未培养物种,表明这些未知物种在跨人群环境中普遍存在。

比较CRC与健康样本的基因组丰度发现,低丰度基因组在两组间表现出更显著的分布差异(log2FC较高),且大多具备统计显著性(p≤0.05)。这一结果提示低丰度微生物可能是CRC相关的机会性病原体,其在疾病判别中的贡献不容忽视。
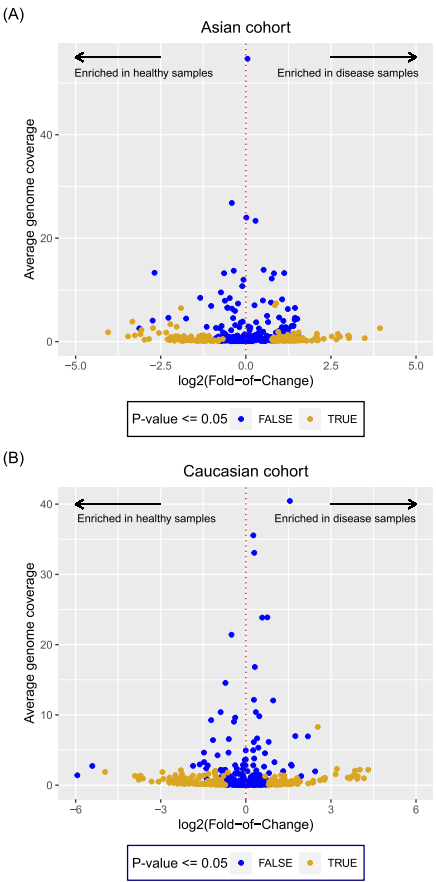
通过随机森林特征重要性排序与累积评估,研究分别从两队列筛选出27和31个关键基因组,其CRC预测AUROC分别达到0.9062和0.9832。这些基因组多数为厚壁菌门(Firmicutes)尤其是梭菌纲(Clostridia),但最高重要性的物种分别为Barnesiella intestinihominis(亚洲)和Prevotella copri(高加索)。值得注意的是,所选基因组中分别有55.56%和70.97%属于低丰度物种,移除它们会导致模型性能下降7%与2%。此外,两队列中分别有2和4个未培养物种被纳入关键基因组集,移除这些未培养物种同样造成预测性能降低(2%与1.3%),突显了未培养物种在疾病预测中的补充价值。
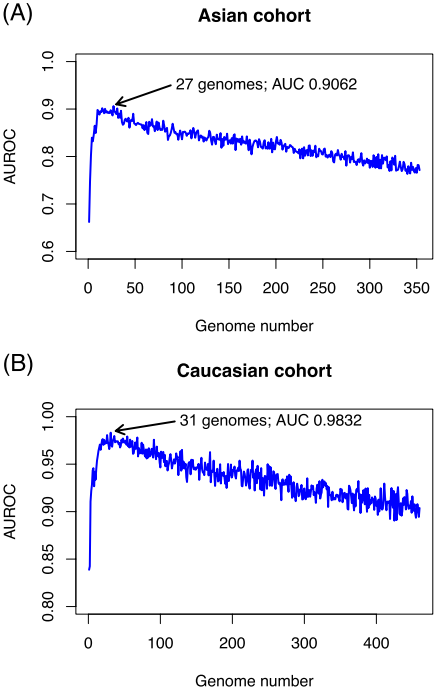

本研究通过宏基因组共组装与分箱技术,成功挖掘了CRC宏基因组数据中的低丰度和未培养物种,证实这些微生物在结直肠癌判别中具有高度预测价值。结果显示,不同人群(亚洲与高加索)中关键物种组成存在明显差异,暗示饮食与文化因素可能塑造了不同的疾病相关菌群结构。方法上,共组装策略显著提高了低丰度物种基因组的回收质量,但也面临计算资源消耗高和菌株混合组装等技术挑战。
值得注意的是,研究中筛选出的关键物种如Barnesiella intestinihominis和Prevotella copri此前已被报道与癌症进展或黏膜炎症相关,与本研究中的富集方向一致。此外,尽管当前方法依赖基因组质量过滤(如MIMAG标准),可能丢失部分未表征物种信息,但整合分箱回收基因组与传统物种注释或许能更全面揭示微生物组与疾病的关联。
该研究不仅强调在疾病微生物组研究中纳入未培养物种的必要性,也为开发基于宏基因组的非侵入性诊断工具提供了一条可行路径。未来研究可进一步结合更先进的特征选择算法(如XGBoost、LASSO等),优化物种筛选流程,推动微生物标记物向临床转化。
生物通微信公众号
知名企业招聘

