
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
Topsicle:基于长读长测序数据估算端粒长度的创新方法及其在跨物种端粒生物学研究中的应用
【字体: 大 中 小 】 时间:2025年09月24日 来源:Genome Biology 9.4
编辑推荐:
本研究针对传统端粒长度测量方法(如TRF、qPCR等)存在通量低、技术要求高且难以应用于特定染色体的问题,开发了Topsicle这一计算工具。该方法利用k-mer分析和变点检测算法,直接从全基因组长读长测序数据中准确估算端粒长度,在拟南芥、玉米和人类癌细胞等数据集上验证了其可靠性,并与TRF等金标准方法高度相关(r=0.74-0.81)。Topsicle无需端粒富集实验或高质量基因组组装,为大规模研究端粒长度变异与衰老、癌症等生命过程的关联提供了高效解决方案。
在生命科学领域,端粒作为染色体末端的保护帽,其长度变异与衰老、癌症和多种疾病密切相关。然而,准确测量端粒长度一直是个技术难题。传统的终端限制性片段(TRF)分析和定量PCR(qPCR)等方法不仅耗时耗力,还受限于通量和准确性。随着长读长测序技术的发展,人们虽然能够获取更长的DNA片段,但如何从海量数据中精准识别端粒区域并准确估算其长度,仍缺乏高效的计算工具。
针对这一挑战,来自堪萨斯大学的研究团队开发了Topsicle这一创新方法,旨在利用全基因组长读长测序数据,通过k-mer分析和变点检测算法,实现端粒长度的高通量、高精度估算。该研究近期发表于《Genome Biology》,为端粒生物学研究提供了强有力的工具。
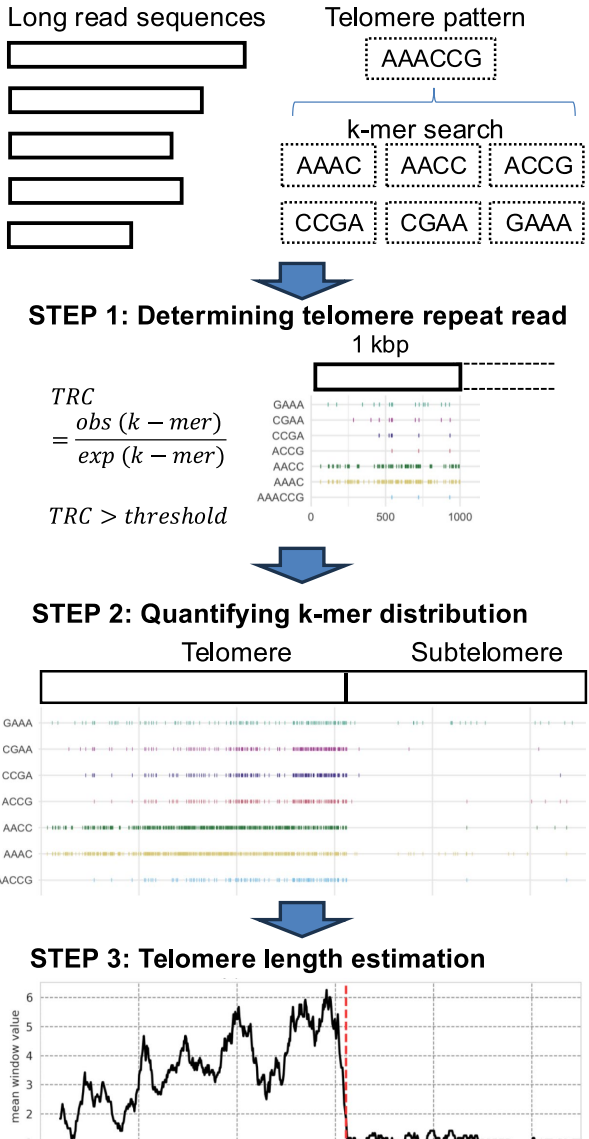
Topsicle的开发基于几个关键步骤:首先,通过k-mer分析识别端粒重复序列;其次,计算端粒重复计数(TRC)统计量以筛选端粒相关读段;最后,应用变点检测算法确定端粒-亚端粒边界,从而估算端粒长度。该方法不依赖于端粒富集实验或完整的基因组组装,使其适用于多种物种和数据类型。
研究团队使用了多个数据集验证Topsicle的性能,包括拟南芥、玉米、 Mimulus 物种以及人类癌症细胞系的测序数据。结果显示,Topsicle在模拟数据和真实数据中均表现出高准确性,与TRF等传统方法的估计结果高度一致。此外,Topsicle还能够处理端粒序列异质性和测序错误,展现出较强的鲁棒性。
在拟南芥中,Topsicle成功估计了104个生态型的端粒长度,发现其长度范围在1720 bp至4720 bp之间,与TRF数据显著相关(r=0.74)。在玉米中,分析了27个基因型,端粒长度变异超过十倍,最短为1010 bp,最长为11745 bp,与TRF结果相关性高达r=0.81。在 Mimulus 物种中,尽管存在端粒序列异质性,Topsicle仍能准确估计长度,与TRF结果几乎一致。在人类癌症细胞系数据中,Topsicle与现有工具Telogator2相比,具有更高的相关性、更低的内存占用和计算时间。
此外,研究还发现,纳米孔测序数据中存在端粒重复序列的特定错误模式,如CCCTAAA被错误识别为CCCTAGG。Topsicle通过使用不同大小的k-mer和相位分析,有效克服了这些错误,确保了长度估计的准确性。
本研究不仅提供了端粒长度估计的新工具,还深入探讨了端粒长度变异在进化、衰老和疾病中的潜在意义。通过大规模数据分析,Topsicle有望促进端粒生物学在生物医学和生态进化研究中的应用,帮助科学家更深入地理解端粒长度与生命史特征之间的关联。
总之,Topsicle作为一个高效、准确的端粒长度估计工具,填补了长读长测序数据分析的空白,为未来研究提供了重要支持。其开源特性将进一步推动端粒研究的发展,助力科学家在更多物种和更广泛条件下探索端粒的生物学功能。
主要技术方法
研究利用Pacific Biosciences(PacBio)和Oxford Nanopore Technologies(纳米孔)长读长测序数据,通过k-mer分析(包括4-mer、5-mer等不同长度)和变点检测算法(binary segmentation)识别端粒重复序列并确定端粒-亚端粒边界。端粒重复计数(TRC)统计量用于筛选端粒相关读段,阈值根据数据特性动态调整。数据分析涵盖拟南芥(104个生态型)、玉米(27个基因型)、Mimulus物种(3个近缘种)及人类癌症细胞系(10个细胞系)的测序数据,与TRF、qPCR等传统方法进行验证比较。
结果
端粒重复序列在染色体末端长读长数据中的空间分布
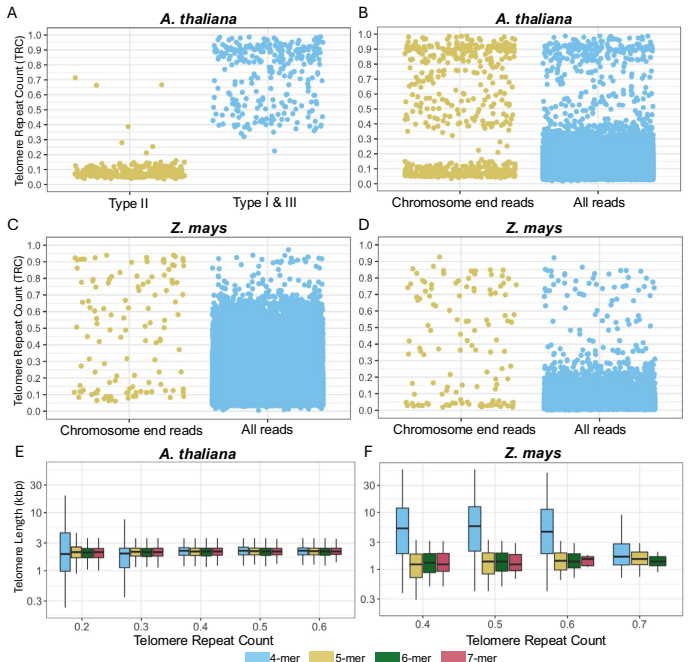
通过分析拟南芥Col-0生态型的纳米孔测序数据,研究发现端粒读段可分为三种类型:I型(高密度端粒重复,可能涵盖端粒-亚端粒边界)、II型(低重复密度,可能来自亚端粒区域)和III型(端粒重复簇中存在间隙,可能因测序错误导致)。这些分布模式为后续端粒长度估算提供了基础。

端粒区域长读长序列中的测序错误分析
研究重点检查了III型读段中的测序错误,发现纳米孔数据中存在偏向性错误,如AAACCCT重复中的CCC同聚物被错误识别为AAACCT,而PacBio数据中错误较少且随机。这种错误模式影响了端粒重复的准确识别,需要通过多相位k-mer分析来校正。
通过k-mer频率总结界定端粒-亚端粒边界
利用滑动窗口(窗口大小100 bp,步长7 bp)和变点检测算法,研究能够准确识别端粒-亚端粒边界。例如,在拟南芥III型读段中,端粒重复k-mer密度在3000 bp处显著下降,边界位置得以确定,端粒长度估计为2900 bp,与实验数据一致。
端粒重复计数(TRC)统计量筛选拟南芥端粒长读段
TRC统计量基于端粒重复k-mer在读段末端的密度计算,能够有效区分端粒相关读段(I型和III型)和非端粒读段(II型)。在拟南芥和玉米数据中,TRC值分布显示双峰模式,通过阈值(如0.4)可筛选端粒读段用于长度估计。

Topsicle性能模拟
通过模拟读段(长度和错误率变化),研究测试了Topsicle在不同条件下的表现。结果显示,在错误率低于5%时,Topsicle估计长度与真实值差异小于100 bp;错误率高达30%时,低估程度小于10%。覆盖度模拟表明,即使在低覆盖(2x)下,Topsicle仍能保持估计稳定性。
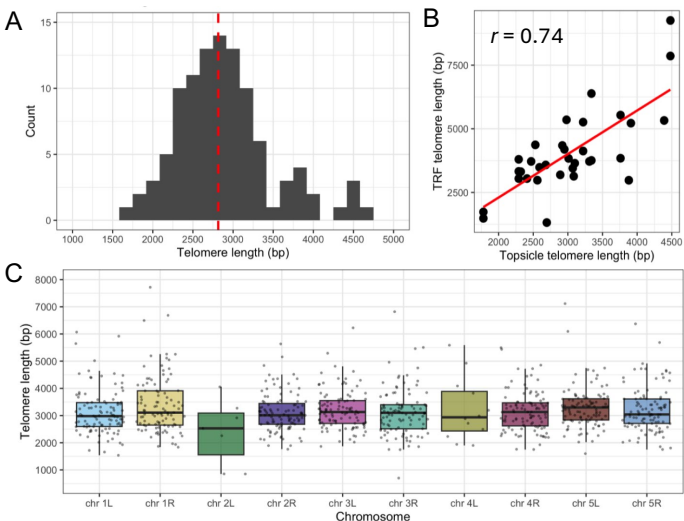
Topsicle在拟南芥长读长测序群体基因组数据集中的应用
应用Topsicle分析了104个拟南芥生态型,端粒长度范围1720-4720 bp,与TRF估计高度相关(r=0.74)。与短读长计算方法(k-Seek、Computel)相比,Topsicle相关性更高(r=0.85)。研究还实现了染色体特异性端粒长度估计,发现各染色体端粒长度无显著差异。
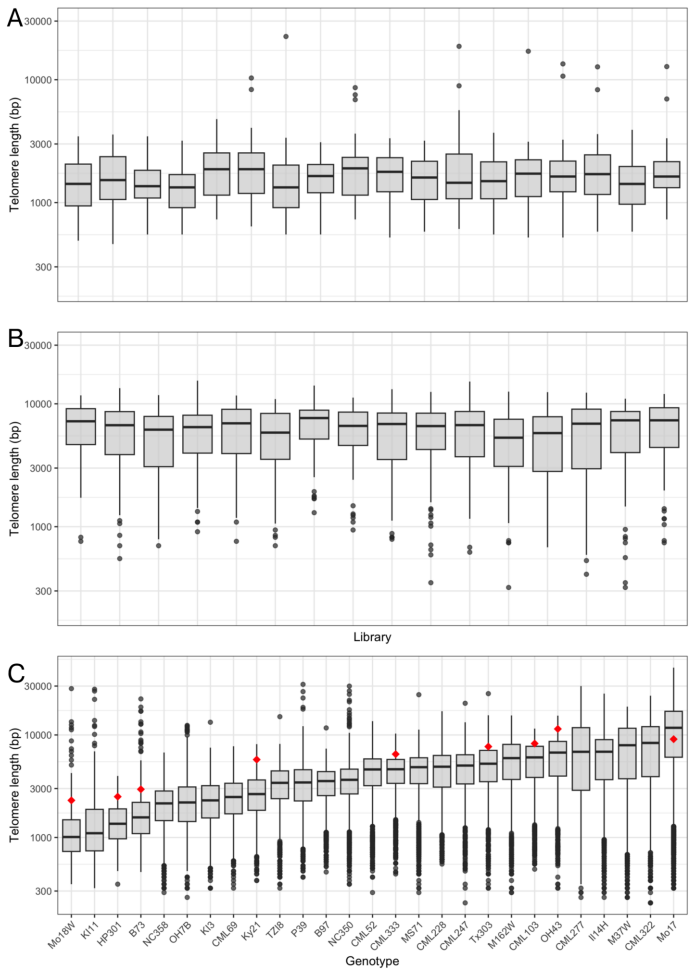
Topsicle在玉米长读长测序数据中的应用
分析了27个玉米基因型,端粒长度变异超过十倍(1010-11745 bp),与TRF估计相关(r=0.81)。研究发现TRF估计较长,因包含亚端粒区域;体外TRF分析证实这一点。不同测序文库间端粒长度无显著差异,表明Topsicle估计的一致性。
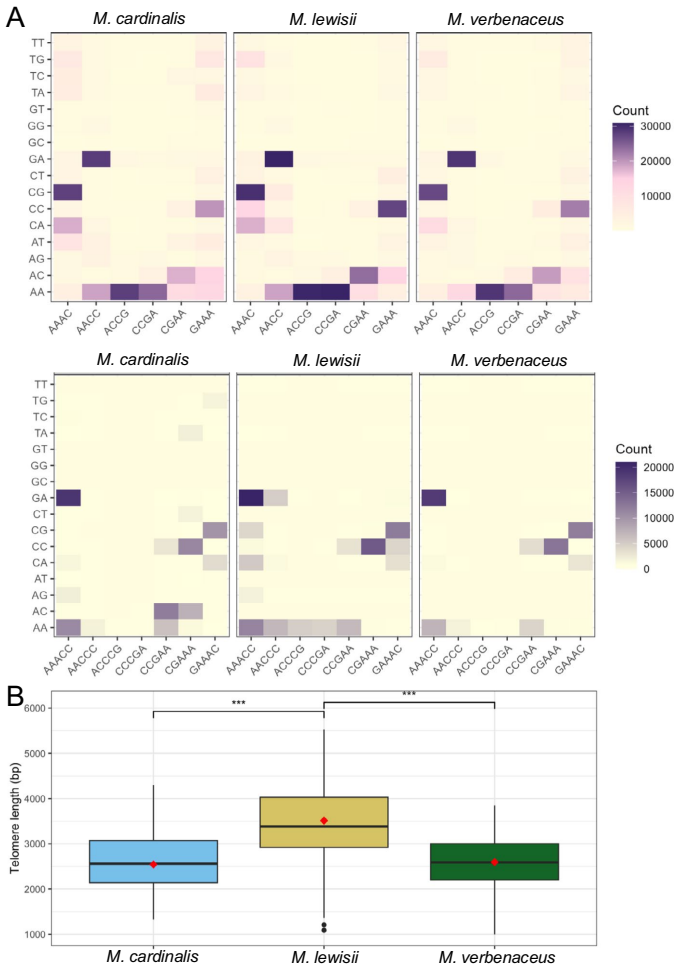
Topsicle在具有序列异质性端粒的Mimulus物种中的应用
在Mimulus物种中,端粒重复序列存在自然变异(如AAACCG和AAACCCG混合),Topsicle通过使用4-mer重复序列和TRC阈值0.4,准确估计了端粒长度(M. cardinalis 2560 bp, M. lewisii 3385 bp, M. verbenaceus 2590 bp),与TRF结果几乎一致。
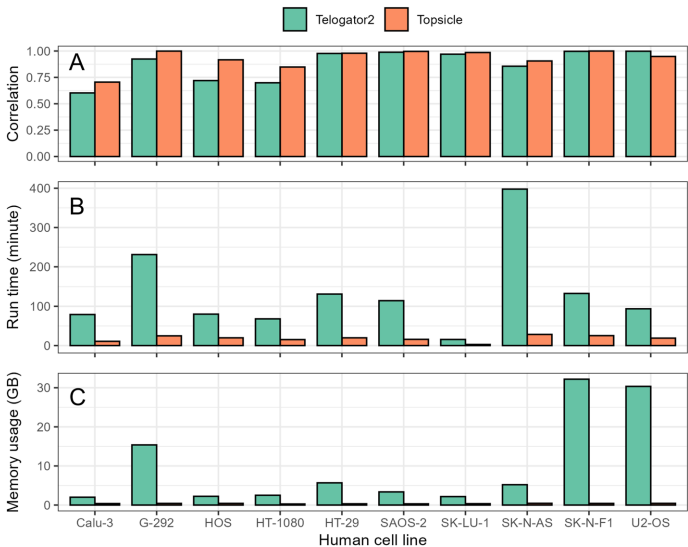
Topsicle在人类基因组学和癌细胞系长读长测序数据中的表现
与Telogator2等工具相比,Topsicle在人类癌症细胞系数据中表现出更高相关性、更低内存占用和计算时间。在10个人类基因组样本中,Topsicle与多种计算方法(k-Seek、Computel、Telseq)估计高度相关(r=0.896-0.944),且未受间质端粒序列影响。
结论与讨论
Topsicle作为一个高效、准确的端粒长度估计工具,填补了长读长测序数据分析的空白。其优势在于无需端粒富集实验或高质量基因组组装,适用于多种物种和数据类型。通过k-mer分析和变点检测算法,Topsicle能够处理测序错误和端粒序列异质性,提供可靠的长度估计。在拟南芥、玉米、Mimulus和人类数据中的验证表明,其与金标准方法高度一致,且适用于大规模群体基因组研究。
该工具的成功开发将促进端粒生物学在生物医学和生态进化研究中的应用,帮助科学家更深入地理解端粒长度变异与衰老、癌症等生命过程的关联。未来工作可进一步优化算法,扩展至更多物种和数据类型,推动端粒研究的发展。
生物通微信公众号
知名企业招聘

